


Riportiamo di seguito la risposta tecnica della Dr.ssa Loretta Bolgan alla critica pubblicata dal Dott. Bucci sulle analisi Corvelva.
Gent.mo dott. Bucci,
di seguito Le rispondo alle criticità da Lei sollevate in particolare nel Suo articolo pubblicato su: CattiviScienziati.com
Innanzitutto vorrei farLe notare che NON è originale l’utilizzo di tecnologie NGS/HTS (Next Generation Sequencing o High-Throughput Sequencing) su substrati complessi di origine biotecnologica, come i vaccini.
La tecnologia NGS è stata già utilizzata in prodotti vaccinali, ad esempio:
- nell’analisi di lotti commerciali del vaccino per il rotavirus (Victoria JG, Wang C, Jones MS, Jaing C, McLoughlin K, Gardner S, et al. Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J Virol 2010;84:6033–40) permettendo il successivo blocco della commercializzazione dei lotti contaminati dal Circovirus Porcino di tipo 1 (PCV1), nonostante in precedenza il lotto avesse superato tutti i test previsti per lo sviluppo, le sperimentazioni cliniche e la produzione (Dubin, G.; Toussaint, J.F.; Cassart, J.P.; Howe, B.; Boyce, D.; Friedland, L.; Abu-Elyazeed, R.; Poncelet, S.; Han, H.H.; Debrus, S. Investigation of a regulatory agency enquiry into potential porcine circovirus type 1 contamination of the human rotavirus vaccine, Rotarix: approach and outcome. Hum. Vaccines Immunother. 2013, 9, 2398–2408).
- nella rilevazione di un nuovo Rhabdovirus nella linea cellulare Sf9 (M H, Galvin TA, Glasner DR, Shaheduzzaman S, Khan AS. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J Virol 2014;88:6576–85)
- nell’identificazione del virus vaccinale della parotite nel cervello di un bambino immunocompromesso, morto per encefalite (Morfopoulou S, Mee ET, Connaughton SM, Brown JR, Gilmour K, Chong WK, Duprex WP, Ferguson D, Hubank M, Hutchinson C, Kaliakatsos M, McQuaid S, Paine S, Plagnol V, Ruis C, Virasami A, Zhan H, Jacques TS, Schepelmann S, Qasim W, Breuer J. Deep sequencing reveals persistence of cell-associated mumps vaccine virus in chronic encephalitis. Acta Neuropathol. 2017 Jan;133(1):139-147. doi: 10.1007/s00401-016-1629-y. Epub 2016 Oct 21. PubMed PMID: 27770235).
Inoltre nel lavoro collaborativo del 2017 ‘A Multicenter Study To Evaluate the Performance of High-Throughput Sequencing for Virus Detection’ (A Khan AS, Ng SHS, Vandeputte O, Aljanahi A, Deyati A, Cassart JP, Charlebois RL, Taliaferro LP. A Multicenter Study To Evaluate the Performance of High-Throughput Sequencing for Virus Detection. mSphere. 2017 Sep 13;2(5).pii:e00307-17. doi: 10.1128/mSphere.00307-17. eCollection 2017 Sep-Oct. PubMed PMID: 28932815; PubMed Central PMCID: PMC5597969), si evidenzia come analisi eseguite con differenti strumenti HTS, con protocolli diversi di preparazione dei campioni e di analisi dati, su campioni mimanti materiali biologici complessi, hanno dimostrato una sensibilità simile nella rilevazione dei virus in 3 diversi laboratori.
Infine, nel ‘Report of the international conference on next generation sequencing for adventitious virus detection in biologicals’ (Khan AS, Benetti L, Blumel J, Deforce D, Egan WM, Knezevic I, Krause PR, Mallet L, Mayer D, Minor PD, Neels P, Wang G. Report of the international conference on next generation sequencing for adventitious virus detection in biologicals. Biologicals. 2018 Sep;55:1-16. doi: 10.1016/j.biologicals.2018.08.002. Epub 2018 Aug 6. PubMed PMID: 30093175), report di una conferenza internazionale tenutasi a Rockville (MD) dal 26 al 27 ottobre 2017, co-organizzata da IABS (International Alliance for Biological Standardization) e dall’FDA (U.S. Food and Drug Administration), a cui hanno partecipato centoventotto scienziati da 16 diversi paesi, tra cui ricercatori Glaxo SmithKline, Merck e Sanofi, si afferma che:
“I principali vantaggi dell’NGS per i test di sicurezza per il rilevamento dei virus sono l’approccio rapido e non mirato, che può essere utilizzato in più substrati e che consente di rilevare un'ampia gamma di virus, comprese varianti e nuove specie; e che non vi è alcuna necessità di amplificazione specifica, mentre la PCR richiede il targeting di una sequenza specifica”
E ancora:
“Le tecnologie HTS stanno rivelando alcuni dei limiti attuali dei programmi per testare materiali biologici e potrebbero integrare lacune in questi programmi e aumentare la sicurezza del prodotto”
E nelle conclusioni del report si legge:
“Continui sforzi collaborativi e scientifici, scambi tra agenzie normative e altri enti pubblici, industria, laboratori accademici e fornitori di servizi faranno avanzare il campo dell’NGS con l'obiettivo di garantire la sicurezza dei prodotti biologici che impattano sulla salute umana e animale”
Rispondo di seguito alle Sue osservazioni, in particolare a quelle che ci permettono di spiegare meglio i dati preliminari che abbiamo prodotto su alcuni lotti di Priorix Tetra.
Osservazione 1
“Ci si chiede come questa pratica di segretezza, contraria ad ogni criterio di condivisione dei dati, si possa conciliare con le continue richieste di trasparenza rivolte dall’associazione CORVELVA e dal presidente dell’Ordine Nazionale dei Biologi in tema di sicurezza vaccinale: è lecito lanciare un allarme, senza fornire tutti i dati necessari alla valutazione della sua consistenza?
Come possono la comunità scientifica ed i cittadini valutare il peso di certe affermazioni, se i dati originali completi sono tenuti segreti, in attesa che un pubblico ministero decida se e come procedere in un’azione giudiziaria?”
Risposta 1
I dati di metagenomica divulgati da Corvelva per il Priorix Tetra (vaccino MPRV) e discussi nel report pubblicato a dicembre erano preliminari (è stata divulgata sul sito Corvelva il 23 gennaio una versione aggiornata, frutto di un ulteriore passo avanti nella analisi dei dati), perché, come già ribadito in diverse occasioni, i dati divulgati finora risultano da un percorso di ricerca e sviluppo tecnologico iniziato nel 2017, con lo scopo di mettere a punto e ottimizzare tutta la procedura, dalla lavorazione del campione fino alla analisi bioinformatica. Si è deciso di divulgare in corso d’opera i risultati, man mano che si rendono disponibili, in assoluta trasparenza (proprio il contrario di quello che viene dichiarato da Lei come ‘pratica di segretezza’!) e comunque fortemente supportati dalla letteratura scientifica sull’argomento.
A nostro avviso, quello che abbiamo divulgato finora su questo vaccino, soprattutto riguardo l’elevata quantità di DNA fetale umano, non è niente di nuovo o inaspettato e tantomeno sconosciuto alle aziende farmaceutiche.
Osservazione 2
“Si può agevolmente osservare come il totale di reads indicato in alto a sinistra (evidenziato in giallo) è pari a oltre 6 milioni; tuttavia, la somma di tutte le reads divise per classe di organismo è pari a circa 5 milioni e mezzo, con un “ammanco” di oltre 700.000 reads (e una somma percentuale, indicata dal box rosso, pari a 88%). Questa semplice addizione, ripetuta su tutte le tabelle, pone la domanda di dove siano finite le reads mancanti (una percentuale non certo trascurabile) e perchè non siano state riportate. Resta chiaramente aperta la possibilità che i ricercatori abbiamo sbagliato le somme”
Risposta 2
Diciamo che 700.000 reads in più o in meno cambiano di poco la sostanza delle analisi di screening preliminare effettuate. Le sequenze mancanti rientrano nel computo delle sequenze ‘unassigned’. Come già detto i dati non sono definitivi e ci possono essere delle imprecisioni nei report. Tutto verrà revisionato e corretto man mano che il lavoro procede.
Osservazione 3
“Il genoma del virus della rosolia, uno dei virus attenuati necessari a conferire l’immunogenicità, non sarebbe presente. La rivelazione a livelli molto bassi, al limite del rumore statistico, di un microorganismo nel campione in esame non dipende quindi dalla sua assenza, ma dal metodo di identificazione non ottimizzato. La stessa, identica difficoltà può incontrarsi per l’identificazione di qualunque altro genoma, per il quale il metodo di analisi non sia stato opportunamente calibrato; la mancata rivelazione del genoma del virus della rosolia (oltretutto un virus a singola elica incapsulato) è ascrivibile quindi al metodo utilizzato, senza la necessità di invocare la sua assenza nei lotti vaccinali esaminati”
Risposta 3
La sua tesi è condivisibile. Siamo in fase di ricerca e sviluppo, però la tecnologia è molto robusta e riconosciuta dalla comunità scientifica che si occupa di deep sequencing per la rilevazione di virus in substrati biologici complessi. Prove inter-laboratorio e l’introduzione di opportuni standard di mix virali certificati ci permetteranno di verificare o smentire l’ipotesi da Lei proposta, cioè che il metodo non è (ancora) ottimizzato per i virus.
In ogni caso il genoma della rosolia è stato poi trovato nel lotto in esame, aumentando la profondità di sequenziamento (114 sequenze paired-end lunghe 125bp, su circa 260 milioni di sequenze prodotte) come descritto nel secondo report di gennaio 2019. Inoltre nel vaccino MMRVax Pro della Merck, analizzato nel 2017 con lo stesso metodo, il genoma del virus della rosolia è stato trovato senza dover andare a profondità così elevate, dimostrando che il metodo funziona.
Osservazione 4
“si rileverebbero i genomi di numerosi organismi contaminanti appartenenti a numerosi taxa, tra cui elminti, batteri e virus patogeni umani avventizi. Vi è poi un’ulteriore problema nelle tabelle presentate, costituito dalla abbondante frequenza di sequenze riconducibili a virus integrati nel genoma umano (endovirus di varia natura) o a sequenze retrovirali note per essere reperibili nel genoma umano normale (questo è il caso ad esempio di sequenze retrovirali integrate che possono essere scambiate per HIV) o nel genoma delle cellule embrionali di pollo usate per la produzione del vaccino, tipicamente riscontrabili come false assegnazioni a genomi contaminanti nel sequenziamento di DNA o RNA a seconda della particolare endosequenza considerata. Innanzitutto, vi sono numerose specie elencate nelle tabelle del report, che sono ritrovate sulla base di un numero di reads pienamente rientrante nel rumore statistico. 3 o meno reads è una procedura azzardata che porta ad un alto numero di falsi positivi”
Risposta 4
La versione del report divulgato sul sito Corvelva a dicembre, a cui Lei si riferisce, è stata aggiornata recentemente con una versione in cui è stata preliminarmente validata una parte dei contaminanti con un software alternativo, e poi manualmente. L’analisi in prima battuta era stata eseguita volutamente senza filtri per evidenziare ogni possibile segnale e tutte le problematiche relative ad una analisi ‘aperta’. Lo scopo del nostro lavoro non è, come ribadito più volte, effettuare un’analisi del rilascio del lotto, ma effettuare uno screening preliminare, e successivamente una conferma inter-laboratorio sulle criticità emerse, mediante l’uso di una tecnologia ben consolidata in ambito genomico, già applicata sui vaccini e di prossimo utilizzo anche da parte delle agenzie regolatorie e dalle grosse aziende farmaceutiche per aumentare la qualità e di conseguenza la sicurezza del prodotto.
Se Lei definisce il limite di 3 reads (sulla base di cosa?), allora potremmo per lo meno pensare probabili i segnali di presenza da retrovirus endogeni in questo vaccino (Human Endogeneous Retrovirus K: 32 reads da 125bp, corrispondenti a 4000bp e HERVH-env 62: 4 reads, corrispondenti a 500bp), nei dati RNA-seq, che per definizione rappresentano il materiale trascritto e quindi potenzialmente attivo.
Concordo invece con Lei sul fatto che le sequenze retrovirali trovate nei dati DNA-seq possono essere provirus integrati nel genoma umano, vista la quantità elevatissima di DNA umano nel vaccino in questione.
Ricordo che nel vaccino Attenuvax (vaccino per il morbillo), 4 sequenze che coprivano 700bp (Victoria JG, Wang C, Jones MS, Jaing C, McLoughlin K, Gardner S, et al. Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. J Virol 2010;84:6033–40) hanno permesso di individuare mediante NGS una contaminazione da virus ALV (Avian Leukosis Virus) per la prima volta in un vaccino, poi confermata mediante PCR nested.
Osservazione 5
“Per quel che riguarda il primo di questi potenziali contaminanti, il batterio azotofissatore non patogeno del suolo Bradyrhizobium, il suo “apparire” nei laboratori di sequenziamento è ben noto come problema legato alla preparazione dei campioni da sequenziare, ascrivibile a contaminazione dei kit di purificazione del DNA, l’acqua e i reagenti da utilizzare. Un problema ben conosciuto, a causa del quale nei lavori di genomica “esplorativa” in genere si sequenzia in parallelo al campione sotto esame un campione di controllo, per esempio contenente acqua”
Risposta 5
I bianchi sono stati fatti (sia di estrazione che di libreria), ma non si sono ottenute librerie e quindi non è stato possibile farli correre sul sequenziatore per verificare esattamente qual’è la contaminazione da reagenti/ambientale. Si stanno facendo comunque sforzi in tal senso per cercare di capire quali sono le contaminazioni ‘fisiologiche’ e anche le prove inter-laboratorio aiuteranno nella determinazione delle possibili contaminazioni batteriche laboratorio dipendenti.
Osservazione 6
“Per quello che riguarda i due tipi di vermi rivelati nell’analisi di sequenziamento di RNA, è sufficiente osservare come essi non siano presenti nelle corrispondenti analisi di sequenziamento del DNA, dove dovrebbero essere molto meglio rilevabili; di conseguenza, l’effettiva presenza delle due specie appare come un artefatto, non confermabile alla luce degli stessi dati di sequenziamento di DNA presentati.
Alla luce delle considerazioni fatte, non è quindi possibile confermare la presenza di nessuno dei genomi contaminanti riportati, ed esistono forti indizi che portano a pensare alla presenza di numerosi falsi positivi”
Risposta 6
La seconda versione del report ha già previsto una prima validazione di alcuni organismi trovati, sia con un software alternativo, che manualmente.
Le valutazione delle errate attribuzioni del software Kraken (peraltro uno tra i software più comunemente utilizzati dalla comunità scientifica per analisi metagenomiche whole-genome) sta servendo per sviluppare una pipeline bioinformatica ad hoc per questo tipo di analisi, che non necessiti dell’utilizzo di altri software e dell’ispezione manuale (peraltro comune nel campo della metagenomica per problemi allo stato dell’arte di assenza di database specifici e particolarmente curati).
Osservazione 7
“Il DNA umano rilevato sarebbe ad alto peso molecolare e la copertura del genoma umano sarebbe totale, cosicchè sarebbe l’intero genoma delle cellule fetali umane e non porzioni di esso ad essere presente nei lotti di vaccino esaminati.
“nel vaccino Priorix Tetra il DNA genomico umano è ad alto peso molecolare (>10.000bp) e la totale copertura in sequenza dell’intero genoma umano di riferimento (HG-19) dimostra che è l’intero genoma delle cellule fetali utilizzate per la coltura dei virus vaccinici ad essere presente e non solo porzioni di esso. “
Nessuna evidenza diretta è data per l’affermazione riportata, fatta salva la presenza di DNA di peso intorno a 100.000 basi rivelato mediante gel elettroforesi dagli autori (documentato con una figura di scarsa qualità nel documento CORVELVA, della quale sarebbe opportuno ottenere un originale ad alta risoluzione per valutare se i dati siano presentati in maniera integra).
Gli autori tuttavia dimenticano che il DNA di alcuni dei virus attenuati presenti nel vaccino in esame è di dimensioni circa pari a quanto da loro rivelato nel gel (per esempio, il DNA del virus della varicella è di 125 kb); pertanto, pur volendo intravedere traccia di DNA ad alto peso nell’immagine di gel presentato, questo può chiaramente essere imputato al DNA dei virus attenuati presenti del vaccino, e non a frammenti di DNA umano. Ancora una volta, siamo di fronte ad una sovrainterpretazione del risultato sperimentale. Per quello che riguarda il secondo elemento su cui si basa l’affermazione di CORVELVA, cioè che sarebbe presente nei vaccini l’intero genoma umano, ricordiamo che non vi è nemmeno bisogno di avere accesso ai dati di copertura dettagliati per pesarne la consistenza: alla profondità di sequenziamento utilizzata e con il numero di reads ottenute, questa affermazione è totalmente implausibile. Inoltre la tecnica di sequenziamento utilizzata, basata su sequenze corte, 125 nucleotidi, non permette di definire se un genoma sia frammentato. La tecnica di sequenziamento utilizzata permette solo di stimare la frazione di DNA genomico sequenziato”
Risposta 7
Il DNA umano in questo vaccino è in rapporto relativo di circa 8 a 1 rispetto al DNA della varicella (88% delle reads è di origine umana, rispetto al 11% della varicella nell’ultimo lotto analizzato A71CB256A). Altri virus dsDNA attenuati non ci sono nel vaccino perché i virus del morbillo, parotite e rosolia sono virus a RNA a singolo filamento che non possono essere visualizzati su gel d’agarosio. L’NGS è una tecnologia quantitativa, perciò una semplice quantificazione fluorimetrica del DNA totale estratto dal vaccino (es. lot. A71CB256A = 3,7 microgrammi per dose), associata alla considerazione di quantificazione relativa fatta sopra (8:1), ci permette di poter dire che il DNA cellulare è di circa 2,9 microgrammi per dose, rispetto a circa 0.74 microgrammi di DNA della varicella. E’ quindi plausibile che almeno una porzione del DNA ad alto peso molecolare che si vede su gel possa essere quello delle cellule della linea cellulare fetale MRC5.
L’evidenza diretta che dentro questo prodotto ci sia un genoma umano COMPLETO (cioè con geni e sequenze non codificanti), frammentato o meno, è data dal risultato dell’allineamento delle reads di derivazione umana sul riferimento umano hg19.
Nella seguente tabella, evidenziato in arancione, è riportato il risultato espresso in ‘Av_cov = copertura media’ dell’allineamento delle sequenze umane dei 3 lotti di Priorix testati (1st, 2nd e 3rd lot) sui cromosomi umani. Nella colonna 1, il chrM è il DNA mitocondriale, mentre i Chr1 fino a ChrY sono i cromosomi umani assemblati, inclusi i cromosomi sessuali X e Y. Nella colonna 2 è riportata la lunghezza dei cromosomi umani assemblati espressa in paia di basi.
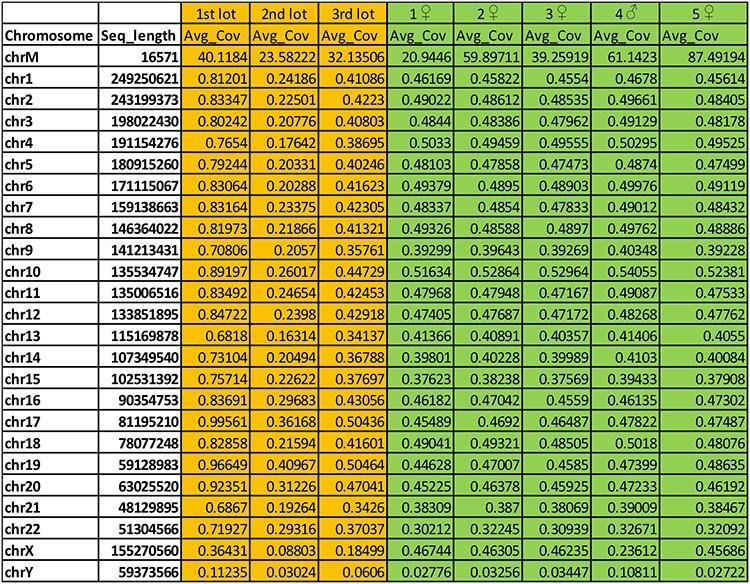
La copertura è bassa (Avg_cov in media lungo ogni cromosoma < di 1x) ma l’omogeneità di distribuzione delle reads che si allineano in modo univoco, lungo tutti i cromosomi umani e la presenza anche di reads che si allineano a più alta copertura sul genoma mitocondriale (nelle cellule umane ci sono 2 genomi, uno nucleare grande circa 3Gbp e diviso in cromosomi e uno mitocondriale più piccolo, circolare, di circa 16Kbp) permette comunque di riconoscere indiscutibilmente una situazione simile ad un low-pass genome sequencing di un genoma umano individuale. Per una più facile comprensione di quanto affermato, nella parte indicata in verde sono riportati dei low pass whole genome sequencing (5 campioni, 4 femmine e 1 maschio) umani ad una profondità analoga a quella prodotta per i 3 lotti vaccinali di Priorix Tetra.
Dal rapporto tra la copertura media per il cromosoma X e Y si può dedurre anche l’indicazione del sesso dell’individuo (molto probabilmente maschile) nel vaccino.
Il DNA umano contenuto nei lotti di Priorix ad ora sequenziati, è stato anche qualificato come appartenente alla linea fetale MRC-5 cioè la linea cellulare continua da tessuto polmonare di un feto abortivo maschile degli anni ’60, in cui vengono fatti crescere il virus della varicella e della rosolia. L’analisi delle varianti del DNA mitocondriale presente nel vaccino rispetto al DNA mitocondriale della linea MRC-5 (il DNA della linea cellulare è stato acquistato presso ATCC, principale risorsa di materiali biologici standard al mondo) evidenzia che sono lo stesso individuo.
Osservazione 8
“vi è da tempo evidenza che anche dosi altissime di DNA umano non sia in grado di indurre rischi significativi neppure su periodi di osservazione lunghi, come confermato da studi su primati non umani, dall’esperienza clinica con i vaccini e altri farmaci biologici e da studi recenti di valutazione del rischio che suggeriscono come anche in quei paesi non europei dove esistono soglie al quantitativo di DNA iniettabile tali soglie siano inutilmente stringenti”
Risposta 8
L’articolo del 1995 (https://www.sciencedirect.com/science/article/pii/S104510568570036X?via%3Dihub) da Lei citato, riguarda uno studio sui primati, che non è a mio avviso certo sufficiente a dimostrare l’assenza di infettività, oncogenicità e autoimmunità del DNA umano iniettato nei bambini, come peraltro dichiarato dagli stessi autori nella parte finale della discussione. Nell’esperimento viene iniettato un grosso quantitativo di DNA umano in scimmie, quindi non DNA della stessa specie.
Il secondo articolo citato (https://www.ncbi.nlm.nih.gov/pubmed/23569076) afferma molto chiaramente che il DNA residuo è infettivo a 2 microgrammi, anche se in ogni caso questo limite viene ricavato da una valutazione statistica e non da dati sperimentali. In ogni caso, seguendo le indicazioni dell’autore, il vaccino Priorix avrebbe una quantità di DNA fetale chiaramente in linea con quella determinata come a rischio infettivo.
La Merck per il vaccino Varivax (varicella, virus vivo attenuato) dichiara nel bugiardino americano la presenza di DNA fetale umano derivato da cellule MRC-5 che dalle analisi eseguite dalla D.ssa Deisher, risulta essere in quantità di circa 2 microgrammi.
Nel caso del bugiardino italiano del Priorix Tetra non viene invece indicata la presenza di DNA della linea MRC-5, pur essendo in quantità dello stesso ordine di grandezza di quello contenuto nel Varivax americano. In ogni caso 2 microgrammi di DNA fetale di MRC-5 non è a mio avviso una quantità residuale, ma un vero e proprio componente del vaccino.
Gli studi in vitro della dott.ssa Deisher, che ci auguriamo saranno pubblicati a breve, purtroppo mostrano risultati sconcertanti quando un vaccino contenente DNA umano fetale (ipometilato) viene incubato in vitro con cellule staminali ematopoietiche umane. Ma attendiamo che la ricerca venga pubblicata e poi ci si potrà riconfrontare sull’argomento.
dott.ssa Loretta Bolgan*
*Dottore in chimica e tecnologie farmaceutiche, con dottorato in scienze farmaceutiche ad Harvard medical school Boston. Ha lavorato nel settore dell’industria farmaceutica dove si è occupata di registrazione e sviluppo di progetti di ricerca in ambito oncologico. Consulente di parte legge 210/92, inquinamento ambientale e malattie professionali, ha partecipato all’ultima Commissione parlamentare d’inchiesta sull’uranio impoverito nel gruppo vaccini. Attuale consulente per l’Ordine Nazionale dei Biologi per la tossicologia dei farmaci e dei vaccini, si occupa anche di nutrizione e terapie complementari.
Download: CORVELVA-d.ssa-Loretta-Bolgan-risponde-a-Bucci.pdf