


Queste ultime analisi sono state possibili grazie al contributo attivo delle associazioni francesi Association Liberté Informations Santé (ALIS), Ligue nationale pour la liberté des vaccinations (LNPLV) e dall’associazione australiana Australian Vaccination-risks Network (AVN) che ringraziamo.
I sequenziatori di nuova generazione sono diventati strumenti d’elezione per analisi approfondite nel campo della biologia e della medicina, soprattutto quella di precisione. Questi strumenti consentono di approcciarsi in maniera nuova e più globale ad una serie di applicazioni come sequenziamento de novo, studi di metagenomica, di epigenomica, sequenziamento del trascrittoma e ri-sequenziamento di genomi.
Quest’ultima applicazione (ri-sequenziamento) è molto diffusa in campo umano sia a scopo di ricerca che diagnostico e consiste nel sequenziamento con tecnologia NGS (Next Generation Sequencing) di un intero genoma individuale allo scopo di mappare mutazioni di singolo nucleotide (SNP, pronuncia ‘snip’), inserzioni e delezioni di sequenze più o meno lunghe avvenute in determinate posizioni del genoma e variazioni nel numero di copie di porzioni di genoma/geni (CNV, Copy Number Variants).
Questa procedura è utile per comprendere i meccanismi di sviluppo di alcune patologie in modo da individuare le direzioni per un futuro trattamento clinico, come ad esempio nel caso del cancro. Con questa metodica infatti il patrimonio genetico di un individuo malato di cancro può essere completamente decodificato nel tessuto normale e in quello tumorale, permettendo di capire cos’è cambiato nel genoma e, se possibile, di intervenire con protocolli mirati.
La procedura di ri-sequenziamento prevede che il DNA di un individuo venga spezzato meccanicamente in frammenti di piccole dimensioni (400-500 paia di basi) e che ai frammenti vengano legati dei tratti di DNA artificiale chiamati adattatori, che permettono di legare i frammenti di DNA umano ad una superficie di vetro sulla quale si esegue poi la lettura delle basi (A, C, G, T). La lettura delle basi del DNA avviene mediante reazioni chimiche di incorporazione di nucleotidi marcati con molecole fluorescenti. I milioni di sequenze (reads) che si ottengono dal sequenziamento avvenuto sulla superficie di vetro, vengono poi mappate sul genoma di riferimento umano con opportuni software e quindi vengono identificate tutte le varianti presenti nel genoma analizzato, rispetto al riferimento.
Questa stessa procedura è stata eseguita sul genoma umano presente in Priorix Tetra lot. n. A71CB256A, genoma appartenente alla linea cellulare MRC-5 (di origine fetale); il lavoro é stato svolto presso un’azienda localizzata negli USA, che routinariamente si occupa di analisi di genomi umani mediante ri-sequenziamento. *
* Il nome del laboratorio che ha eseguito questa analisi verrà inserito nel prossimo esposto che depositeremo presso la Procura della Repubblica di Roma nonché agli enti controllori italiani ed europei. Le realtà che stanno depositando i risultati delle analisi finanziate da Corvelva verranno immediatamente aggiornate anche di questi sconcertanti risultati. Non neghiamo di essere, come genitori in primis, angosciati dai risultati che riportiamo di seguito - se già non bastasse quanto scoperto sinora.
Risultati
Il genoma umano di riferimento è risultato essere coperto da reads originate dal DNA vaccinale per il 99.76%, quindi per quasi tutta la sua interezza. Il DNA umano fetale rappresentato in questo vaccino è quindi un genoma individuale completo ovvero è presente nel vaccino DNA genomico di tutti i cromosomi di un individuo di sesso maschile (e infatti il feto da cui deriva la linea cellulare MRC-5 è maschile).
Di seguito vengono riportati i risultati dell’analisi dei vari tipi di varianti rispetto al genoma di riferimento umano.
Varianti a singolo nucleotide (SNP) e corte inserzioni/delezioni (InDels)
Le varianti di singole basi del DNA (SNP, pronuncia ‘snip’) sono polimorfismi, cioè variazioni del materiale genetico, a carico di un unico nucleotide. Le ‘InDels’ sono invece piccole inserzioni e delezioni di meno di 50 pb di lunghezza e costituiscono un'altra classe di varianti genomiche nel genoma umano.
Sono stati identificati nel genoma umano vaccinale un totale di circa 3.6 milioni di SNP (di cui il 98.31% già riportati nel database pubblico dbSNP e 61.805 nuovi, ovvero originali di questo DNA) e circa 804 mila InDels (di cui l’89.42% già riportate in dbSNP e 85.106 nuove).
La quantità di SNP è in linea con quanto riportato in letteratura in un “genoma umano tipico” mentre le indels risultano in quantità superiore rispetto a quanto riportato dal “The 1000 Genomes Project Consortium” ovvero 800 mila rispetto a 600 mila.
CNV (Copy Number Variants) e SV (Structural Variants)
Le varianti in numero di copie (CNV) sono varianti genomiche dovute a variazioni nel numero di copie di frammenti relativamente grandi (più lunghi di 50 bp) tra genomi individuali. Esistono due tipi di CNV: tipo “gain” (guadagno di copie) e tipo “loss” (perdita di copie). Sono state rilevate nel genoma umano vaccinale 218 CNVs di cui 82 di tipo “gain” (che coprono una porzione di genoma complessivamente pari a circa 6.9 milioni di paia di basi) e 136 CNVs di tipo “loss” (che coprono una porzione di genoma di circa 70 milioni di basi).
Come descritto dal The 1000 Genomes Project Consortium in “A global reference for human genetic variation (Nature, vol. 526, 10 Ott. 2015)” un tipico genoma umano contiene da 2.100 a 2.500 varianti di grandi dimensioni tra le quali:
- 1.000 grandi delezioni
- 160 varianti in numero di copie (CNVs)
- 10 inversioni
che influiscono complessivamente, considerando anche le inserzioni, su 20 milioni di basi di sequenza.
Così come constatato per le INDELs corte, anche nel caso delle inserzioni e delezioni di grandi dimensioni il genoma vaccinale risulta quindi essere non in linea con un genoma umano ‘normale’, essendo molto più “riarrangiato” rispetto ad un genoma di un comune individuo.
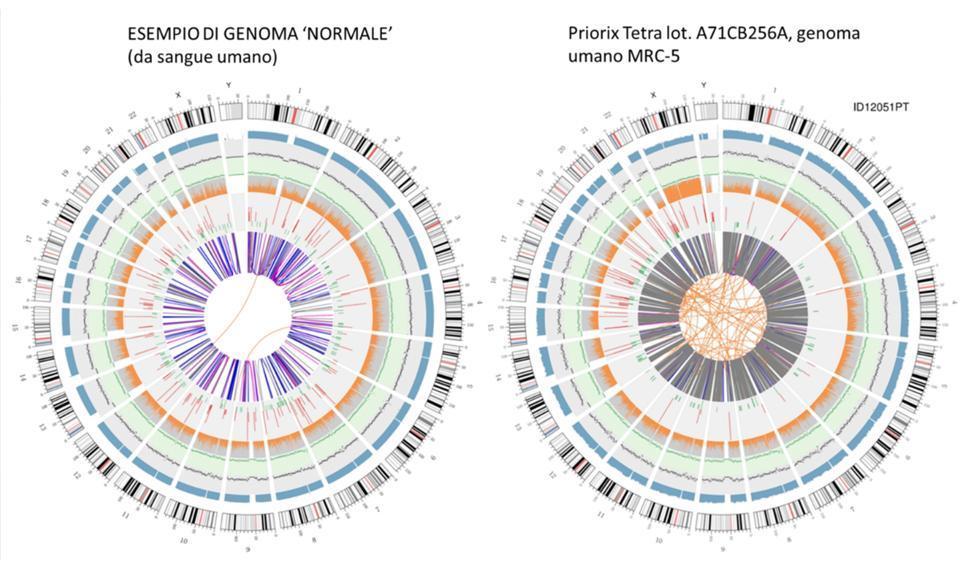
Visualizzazione circolare del genoma (circos plot)
Una rappresentazione grafica del genoma vaccinale chiamata “circos plot” (che si utilizza comunemente per rappresentare un genoma ri-sequenziato), è riportata di seguito, a fianco di un’altra che rappresenta un genoma ri-sequenziato a partire da DNA estratto da sangue di un individuo sano - genoma “normale”:
Significato dei vari cerchi concentrici
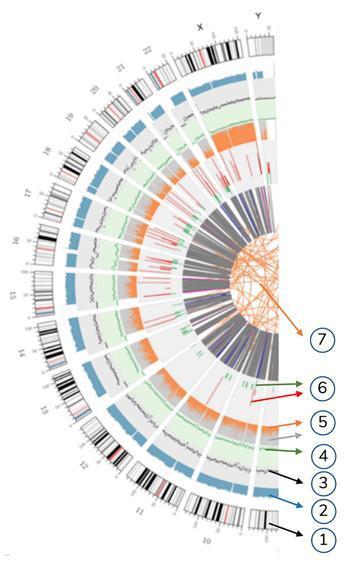
7) L’anello più centrale rappresenta l’inferenza di SV (Varianti Strutturali) nelle regioni esoniche e di splicing. TRA (arancione, traslocazioni), INS (verde, inserzioni), DEL (delezioni, grigio), DUP (duplicazioni, rosa) e INV (inversioni, blu).
6) Il sesto anello rappresenta l’inferenza di CNV (Varianti in numero di copie). Il rosso significa guadagno di pezzi di DNA e il verde significa perdita.
5) Il quinto anello rappresenta la proporzione di SNP in omozigosi (arancione) e in eterozigosi (grigio) in stile istogramma.
4) Il quarto anello (verde) rappresenta la densità snp in stile “grafico di dispersione”.
3) Il terzo anello (nero) rappresenta la densità delle INDELs in stile “grafico di dispersione”.
2) Il secondo anello (azzurro) rappresenta la copertura di reads in stile istogramma.
1) Il cerchio esterno (il primo cerchio) è il numero del cromosoma.
Una comparazione approssimativa può essere fatta anche tra il DNA fetale e il DNA delle cellule HeLa , la linea cellulare immortalizzata utilizzata anche nella produzione del vaccino contro la poliomielite.
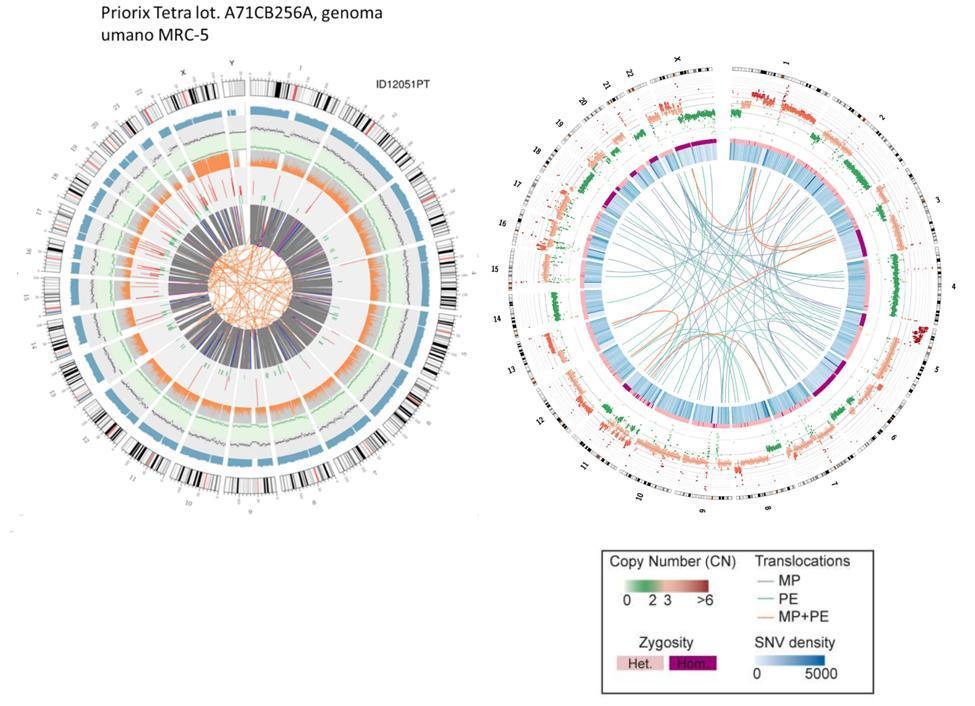
Da sottolineare che le traslocazioni delle cellule HeLa rappresentate nel circos plot dalle linee del nucleo, sono riferite all’intero genoma (quindi parte codificante e non codificante), mentre nel caso delle cellule fetali vaccinali sono riferite solo ai geni codificanti.
Non occorre essere degli scienziati per capire dai circos, semplicemente a colpo d’occhio, che il genoma vaccinale non è un genoma che si può definire “normale”. Le linee arancio intrecciate al centro del circos, non presenti così numerose nell’anello corrispondente del genoma “normale”, fanno già da sole intuire l’anomalia di questo genoma.
Conclusioni
Il DNA genomico umano contenuto nel vaccino Prorix lot. n. A71CB256A è evidentemente anomalo, presentando importanti incongruenze rispetto a un genoma umano tipico, cioè a quello di un individuo sano. Numerose sono le varianti sconosciute (non annotate in database pubblici) e diverse di esse sono localizzate in geni coinvolti nel cancro. Quello che è anche evidentemente anomalo, è l’eccesso di genoma che mostra cambiamenti del numero di copie (CNV) e varianti strutturali (SV), quali traslocazioni, inserzioni, delezioni, duplicazioni e inversioni, molte delle quali coinvolgenti geni.
Il potenziale contributo delle moltissime varianti (non presenti nella letteratura scientifica e nei database pubblici) al fenotipo delle cellule utilizzate per la crescita dei virus vaccinali, non è conosciuto.
All'interno del PDF troverete tutti gli approfondimenti legati anche alle implicazioni per la salute e la corrispondenza con l'EMA.
Download: CORVELVA-Ri-sequenziamento-genoma-umano-Priorix-Tetra.pdf